Mapping reads
We will now map our trimmed reads to a reference. We will use as a reference the bait sequence Ref.fna that is inside the Targeted_enrichment repository we cloned from GitHub. Let’s take a look inside this file:
less /your/path/to/Targeted_enrichment/Ref.fna
>comp39600_c0_seq2
CGGGCTTTAAGAGGGACCTTCTTGACAATGGAAAGCCTACAAGCCTCCTCCATATTTAAGAGGGACCTTTTTGATAAAATAAAACATATATAAATCCACTTAATCCAGTGAATACAGATGAAGAGTCAACCTAGGCTCAACCTGGGCTTCAAGAGGGACCTTCTTGGCAACGGAAAGTCCATGAGCCTCCTCCATATTCAAGTCATGCCCCTCCATGTTCCCAGCTAGCTGCCACCTGAACCCATGAACGAGATTCGCCAAACTTGATTGAATCACCCTAAGCCCGAGAGGATAGCCCGGGCACATTCTTCTTCCAGCCCCAAATGGCAACAACTCAAAGTCATGACCGTTCACATCAATCTCCTTAGTCGGGCCATCAATGAACCTCTCGGGCTTAAACTCATTGGGCCAGCCCCAAACTGTCGGGTCTCTCCCTATGGCCCACACATTCAAAATCACTTGGGTGCCTTTGGGAACGTCATAGCCATCAATTTTACAGTGCTCCCGGGCTTCCCTTGGGATCAGTGTGGGTGCCACAGGGTGTAACCTCATTGTTTCTTTAACAATTGCTTTAATGTAAGGTAGATTGACCATGTCTTGTTCTTCAACCCATCTGTTTCTACCTACCACATTGTCTAGCTCTTGTTGTGCCTGCTTCAAGACTTGTGGCTTTTTAAGAAGCTCTGACATTGCCCATTCCACTGTTATAGAAGAGCTCTGCGTCCCCCCTATGAGAAGATCCTGAGTAATTGCTTTCACATTATGCTTTTGAAGCTTGACCTCGTGGCTAGGATCATCTGCAGCTTGCAAAAGGAGATCCACCATATCCCTGGCAACATAATTCTTAATCCCTCTTCTTCTGTCATTGTGTTCATTCAATATATGTTCCAAAAACCTCTCAAATTTCTTACTCACGACCTTCATCCTCTTCACATATCCCTGCAAATCCAACAATTCAAGCCAAGGAATGAAATCCCCAATATTCAGAAACCCACTATTCAATAACAGAAACTCATCTAGCATCCACTTGAACTCATCCGGCGTAATAACAGCATTTTCGGTTTTGTTAACGTATGTCTTCCCCAAAACCATGCGACTTATGATGTTGAGGTTGAAAAGGTTGAGGTAGTCCTTTAAGAATATGGTTTTGCCCGAAGAACTGTACAGTTGATTTGCCATGATTCTGATCTCTTCCTTTCTGATATACTCATAGGACTCCAAGCTTTTGGGAGAGAAGAGCTCGGTAAAACAGAGTTTGCGGGCTTGGCGCCAGTATGGGGAGTATGGGGCCCAGATAATGTTGGAGTGGTCGAAACTTGTGTACTTACCGGCCGCAGTTCTCGGCCGCGACGCTAAGGAGCCGTCGTGGGTTTGCAGAAAGGCCTTAGCCATTTTCGCTGAGGAGCCAACGACGACAGACTTTGAGCCTAACCAGAGGTGCATGACGGGTCCGTACTTTTGGGATAGAACATGGAGGGACTTGTGGGGGAATAGGCCTATGAGGTTGAGGTTTCCGA
>comp39985_c0_seq4
TTTTTTTATTGTAAAGGGGAGATTTAGTATTGCATTATTTCTTCCATTGTCAAAATGCAAAGGATTACAAGGATCTACAGCTTCGTAGCTGCTTCCATGAAATGTTTCAGTGTTTGGCACTGAAAACAATTCGAATATTTATTTAAAACAAACCAAACTCATTAAAGGCATACATATGTTGGTTAAAAAAAAGAAAAAAAAAAGGCATACATAAAATACACCTCTAAAGAATGCCAATGTTTGATCTAGCACCCTGCATAGTGGCCATCTTCACCCCTCAAAAAGCACTCAATTCGTTCTAGCGCCTCAATAGCAATGGTAGTCGGTGAGATATCTGCAACATCTCTACAGCCCAGTTTTGCAGCAATATTTTCTAAGGACACTCGAGCATTGGCATTGGCATAAGCTTCGCCTCTTTCTTCGAAAAGTGTAGACAAACGCATCAAAGCCCGCTTGTACGCATCTCCTGCTTCATAATGCAGAAGTGGACGAGAATTAGTTCCCACAGCTGCAATTCTCCTGGCCAGGGCTTCTAAAGGTACATCCAACCAAACAGTGATCCCCTTGTGCATATATTTCCAATTGATGGGCCTCACAACTGCACCTCCACCAGTAGAAATTACAAGTCCATGCATAAGTGACAGATTACGCAATACCTCAGTCTCCTTGTCCCTGAAGAAATCCTCTCCATAGTGCTTGAATATATCAGCTACAGAAGTTCCACCAACCTCATGCTCCACCAAGGCATCACTATCAAAAAAGGAATAACCAAGCACTTGTGACAAAATCTTCCCCACAGTTGTCTTCCCTGACCCCATCATTCCAACAAGATATATACAGCGTCCATTTAAATAAGGCTCGATCTCTTGTGTCCTAGTCTGTAACATTAACTCTTCATCAGGAGGGGCCTCTAAGCTTCCAGAGTCCAATGTCAAAGCTGGCAAATTGTCGTAGGAACAAGAAACCTCCAAAGGGGCCGCCCGCCCCCGATTGCGGGAAGGTCTTGCAGTTGCAGGCTGCAAAACACAAATCCGAAGCTTCTGCGGCTCTTCAAAAACTCTAGAGAATTTCACAGAACCACTTAATTTCCTCCCAATCTTCTCGTAATGAATCAATGTTGAGAATTGCAGCCTCTGAGCCGCTTTTGCATCCATGGCGTCGCCGCAAAATAACTGACAACCAGCGTGATATAGCAGTGAACGCAGAGTTTTCAGCACATATACTCAATCCAGATTGACGAGCAGAAAGAAATAAAACGAGAAAATTAATGAACGAAACAGCTCCAGAAATAGGCACGTAAAAG
This is the sequence of our baits. It has the bait name (e.g. >comp39600_c0_seq2 followed by the sequence itself). We can count how many baits we have in this file by counting the number of > since each bait name starts with this symbol:
cat /your/path/to/Targeted_enrichment/Ref.fna | grep -c ">"
So we have 264 loci in our bait set. To map our trimmed reads to the bait set we will use the script bam_me.sh.
bam_me.sh
#! /bin/bash -x
# Catherine Kidner 28 Oct 2014
# Assumes all your reads are in a folder called trimmed and all are gzipped.
echo "Hello world"
acc=$1
echo "Working on $1"
score=G,20,8
fwd_p=${acc}_trimmed_1.fastq.gz
rev_p=${acc}_trimmed_2.fastq.gz
un_p=${acc}_trimmed_1u.fastq,${acc}_trimmed_2u.fastq
sam=${acc}.sam
index=${acc}_sorted.bam
pileup=${acc}.pileup
vcf=${acc}.vcf
bowtie=${acc}_bowtie_output
sorted=${acc}_sorted
bowtie2 --local --score-min $score -x ./Inga_unique_baits -1 $fwd_p -2 $rev_p -U $un_p -S $sam 2>$bowtie
samtools view -bS $sam | samtools sort -o $index $sam
samtools index $index
samtools mpileup -E -uf Ref.fna $index > $pileup
bcftools call -c $pileup > $vcf
rm *.sam
rm *.pileup
exit 0
We can see that this script uses two different software: Bowtie2 and Samtools.
Bowtie2 - extracts from http://bowtie-bio.sourceforge.net/bowtie2/index.shtml:
Here you can see extracts from the Bowtie 2 manual, available at http://bowtie-bio.sourceforge.net/bowtie2/index.shtml.
Bowtie 2 is an ultrafast and memory-efficient tool for aligning sequencing reads to long reference sequences. It is particularly good at aligning reads of about 50 up to 100s or 1,000s of characters to relatively long (e.g. mammalian) genomes. Bowtie 2 indexes the genome with an FM Index (based on the Burrows-Wheeler Transform or BWT) to keep its memory footprint small: for the human genome, its memory footprint is typically around 3.2 gigabytes of RAM. Bowtie 2 supports gapped, local, and paired-end alignment modes.
Bowtie 2 outputs alignments in SAM format, enabling interoperation with a large number of other tools (e.g. SAMtools, GATK) that use SAM. Bowtie 2 is distributed under the GPLv3 license, and it runs on the command line under Windows, Mac OS X and Linux and BSD.
Bowtie 2 is often the first step in pipelines for comparative genomics, including for variation calling, ChIP-seq, RNA-seq, BS-seq. Bowtie 2 and Bowtie (also called “Bowtie 1” here) are also tightly integrated into many other tools, some of which are listed here.
THE bowtie2 ALIGNER
bowtie2 takes a Bowtie 2 index and a set of sequencing read files and outputs a set of alignments in SAM format.
“Alignment” is the process by which we discover how and where the read sequences are similar to the reference sequence. An “alignment” is a result from this process, specifically: an alignment is a way of “lining up” some or all of the characters in the read with some characters from the reference in a way that reveals how they’re similar. For example:
Read: GACTGGGCGATCTCGACTTCG
||||| |||||||||| |||
Reference: GACTG--CGATCTCGACATCG
Where dash symbols represent gaps and vertical bars show where aligned characters match.
End-to-end alignment versus local alignment
By default, Bowtie 2 performs end-to-end read alignment. That is, it searches for alignments involving all of the read characters. This is also called an “untrimmed” or “unclipped” alignment.
When the --local option is specified, Bowtie 2 performs local read alignment. In this mode, Bowtie 2 might “trim” or “clip” some read characters from one or both ends of the alignment if doing so maximizes the alignment score.
Scores: higher = more similar
An alignment score quantifies how similar the read sequence is to the reference sequence aligned to. The higher the score, the more similar they are. A score is calculated by subtracting penalties for each difference (mismatch, gap, etc.) and, in local alignment mode, adding bonuses for each match.
--score-min Sets a function governing the minimum alignment score needed for an alignment to be considered “valid” (i.e. good enough to report). This is a function of read length. For instance, specifying L,0,-0.6 sets the minimum-score function f to f(x) = 0 + -0.6 * x, where x is the read length. See also: setting function options. The default in --end-to-end mode is L,-0.6,-0.6 and the default in --local mode is G,20,8.
from: https://github.com/BenLangmead/bowtie2/blob/master/MANUAL
Samtools: extracts from http://www.htslib.org
Here you can see extracts from Samtools manual, available here http://www.htslib.org.
Samtools is a suite of programs for interacting with high-throughput sequencing data. It consists of three separate repositories:
Samtools: Reading/writing/editing/indexing/viewing SAM/BAM/CRAM format
BCFtools: Reading/writing BCF2/VCF/gVCF files and calling/filtering/summarising SNP and short indel sequence variants
HTSlib: A C library for reading/writing high-throughput sequencing data
Samtools and BCFtools both use HTSlib internally, but these source packages contain their own copies of htslib so they can be built independently.
Samtools is a set of utilities that manipulate alignments in the SAM (Sequence Alignment/Map), BAM, and CRAM formats. It converts between the formats, does sorting, merging and indexing, and can retrieve reads in any regions swiftly.
samtools view -bS $sam | samtools sort -o $_sorted.bam $sam
view: With no options or regions specified, prints all alignments in the specified input alignment file (in SAM, BAM, or CRAM format) to standard output in SAM format (with no header). -b Output in the BAM format. -S Ignored for compatibility with previous samtools versions. Previously this option was required if input was in SAM format, but now the correct format is automatically detected by examining the first few characters of input.
sort: Sort alignments by leftmost coordinates, or by read name when -n is used. An appropriate @HD-SO sort order header tag will be added or an existing one updated if necessary. -o FILE Write the final sorted output to FILE, rather than to standard output.
samtools index $index
index: Index a coordinate-sorted BGZIP-compressed SAM, BAM or CRAM file for fast random access. Note for SAM this only works if the file has been BGZF compressed first.
samtools mpileup -E -uf Ref.fna $index > $pileup
BCFtools is a set of utilities that manipulate variant calls in the Variant Call Format (VCF) and its binary counterpart BCF. All commands work transparently with both VCFs and BCFs, both uncompressed and BGZF-compressed.
mpileup: Generate VCF or BCF containing genotype likelihoods for one or multiple alignment (BAM or CRAM) files. This is based on the original samtools mpileup command (with the -v or -g options) producing genotype likelihoods in VCF or BCF format, but not the textual pileup output. The mpileup command was transferred to bcftools in order to avoid errors resulting from use of incompatible versions of samtools and bcftools when using in the mpileup+bcftools call pipeline.
Generate VCF, BCF or pileup for one or multiple BAM files. Alignment records are grouped by sample (SM) identifiers in @RG header lines. If sample identifiers are absent, each input file is regarded as one sample. -E, –redo-BAQ Recalculate BAQ on the fly, ignore existing BQ tags -uF -f, –fasta-ref FILE The faidx-indexed reference file in the FASTA format. The file can be optionally compressed by bgzip.
Samtools mpileup can still produce VCF and BCF output (with -g or -u), but this feature is deprecated and will be removed in a future release. Please use bcftools mpileup for this instead.
bcftools call -c $pileup > $vcf`
This command replaces the former bcftools view caller. Some of the original functionality has been temporarily lost in the process of transition under htslib, but will be added back on popular demand. The original calling model can be invoked with the -c option.
First, we need to index the bait set for Bowtie2:
bowtie2-build Ref.fna Inga_unique_baits
Note
Here the name Inga_unique_baits is a choice of the user. It will be the name of the indexed baits that Bowtie2 will use for the analysis. You can choose any name of your preference but remember to change accordingly in the bam_me.sh script, more specifically in the line: bowtie2 --local --score-min $score -x ./Inga_unique_baits -1 $fwd_p -2 $rev_p -U $un_p -S $sam 2>$bowtie. Ref.fna is the fasta file containing the bait sequence, we will still need it in a later samtools step.
Now we are ready to run the bam_me.sh script in a loop:
while read f; do ./bam_me.sh "$f" ; done < acc
Tip
In case you are doing your anaylisis in Crop Diversity HPC, you can run this step as an array. An array job is a job in which the script is run concomitantly for each sample and it will be much quicker. In this link you can find more about array jobs: https://help.cropdiversity.ac.uk/slurm-overview.html#array-jobs. Below an example of how to run the bam_me.sh script as an array:
#!/bin/bash
# Assumes all your reads are in a folder called trimmed and all are gzipped.
# Catherine Kidner 28 Oct 2014
# Adjusted for an array job in Mar 2022, Flavia Pezzini
#SBATCH --job-name="bam"
#SBATCH --export=ALL
#SBATCH --mail-user=youremail@yourdomain # enter your email to receive a message once it is done.
#SBATCH --mail-type=END,FAIL
#SBATCH --output ./slurm-%x-%A_%a.out # %x gives job name, %A job ID, %a array index
#SBATCH --partition=short
#SBATCH --cpus-per-task=4 #this is the number of threads, not cores
#SBATCH --mem=2G #adjust this according to your data.
#SBATCH --array=0-4 # the number of samples you have. We have five accessions we use 0-4 because Bash array is zero-indexed (instead of 1-5). It is a good practice to ask for a maximum of 25 tasks at a time.
acc=$(sed -n "$SLURM_ARRAY_TASK_ID"p /path/to/my/acc/file)
echo "Hello world"
echo "Working on $acc"
score=G,20,8
fwd_p=${acc}_trimmed_1.fastq.gz
rev_p=${acc}_trimmed_2.fastq.gz
un_p=${acc}_trimmed_1u.fastq,${acc}_trimmed_2u.fastq
sam=${acc}.sam
index=${acc}_sorted.bam
pileup=${acc}.pileup
vcf=${acc}.vcf
bowtie=${acc}_bowtie_output
sorted=${acc}_sorted
bowtie2 --local --score-min $score -x ./Inga_unique_baits -1 $fwd_p -2 $rev_p -U $un_p -S $sam 2>$bowtie
samtools view -bS $sam | samtools sort -o $index $sam
samtools index $index
bcftools mpileup -E -f Baits_all_loci.fna $index > $pileup
bcftools call -c $pileup > $vcf
rm ${acc}.sam
rm ${acc}.pileup
exit 0
To submit just type: sbatch bam_me_array.sh
In our folder we now have:
FG113 FG35_1.fastq.gz FGIntype_2.fastq.gz KGD465_1.fastq.gz fastqctrimfile
FG113.empties FG35_1_fastqc.html FGIntype_2_fastqc.html KGD465_1_fastqc.html more_tidying.sh
FG113.vcf FG35_1_fastqc.zip FGIntype_2_fastqc.zip KGD465_1_fastqc.zip renaming.sh
FG113_1.fastq.gz FG35_2.fastq.gz FGIntype_bowtie_output KGD465_2.fastq.gz zygia917
FG113_1_fastqc.html FG35_2_fastqc.html FGIntype_forward_paired.fq.gz KGD465_2_fastqc.html zygia917.empties
FG113_1_fastqc.zip FG35_2_fastqc.zip FGIntype_forward_paired_fastqc.html KGD465_2_fastqc.zip zygia917.vcf
FG113_2.fastq.gz FG35_bowtie_output FGIntype_forward_paired_fastqc.zip KGD465_bowtie_output zygia917_1.fastq.gz
FG113_2_fastqc.html FG35_forward_paired.fq.gz FGIntype_forward_unpaired.fq.gz KGD465_forward_paired.fq.gz zygia917_1_fastqc.html
FG113_2_fastqc.zip FG35_forward_paired_fastqc.html FGIntype_forward_unpaired_fastqc.html KGD465_forward_paired_fastqc.html zygia917_1_fastqc.zip
FG113_bowtie_output FG35_forward_paired_fastqc.zip FGIntype_forward_unpaired_fastqc.zip KGD465_forward_paired_fastqc.zip zygia917_2.fastq.gz
FG113_forward_paired.fq.gz FG35_forward_unpaired.fq.gz FGIntype_reverse_paired.fq.gz KGD465_forward_unpaired.fq.gz zygia917_2_fastqc.html
FG113_forward_paired_fastqc.html FG35_forward_unpaired_fastqc.html FGIntype_reverse_paired_fastqc.html KGD465_forward_unpaired_fastqc.html zygia917_2_fastqc.zip
FG113_forward_paired_fastqc.zip FG35_forward_unpaired_fastqc.zip FGIntype_reverse_paired_fastqc.zip KGD465_forward_unpaired_fastqc.zip zygia917_bowtie_output
FG113_forward_unpaired.fq.gz FG35_reverse_paired.fq.gz FGIntype_reverse_unpaired.fq.gz KGD465_reverse_paired.fq.gz zygia917_forward_paired.fq.gz
FG113_forward_unpaired_fastqc.html FG35_reverse_paired_fastqc.html FGIntype_reverse_unpaired_fastqc.html KGD465_reverse_paired_fastqc.html zygia917_forward_paired_fastqc.html
FG113_forward_unpaired_fastqc.zip FG35_reverse_paired_fastqc.zip FGIntype_reverse_unpaired_fastqc.zip KGD465_reverse_paired_fastqc.zip zygia917_forward_paired_fastqc.zip
FG113_reverse_paired.fq.gz FG35_reverse_unpaired.fq.gz FGIntype_sorted.bam KGD465_reverse_unpaired.fq.gz zygia917_forward_unpaired.fq.gz
FG113_reverse_paired_fastqc.html FG35_reverse_unpaired_fastqc.html FGIntype_sorted.bam.bai KGD465_reverse_unpaired_fastqc.html zygia917_forward_unpaired_fastqc.html
FG113_reverse_paired_fastqc.zip FG35_reverse_unpaired_fastqc.zip FGIntype_trimmed_1.fastq KGD465_reverse_unpaired_fastqc.zip zygia917_forward_unpaired_fastqc.zip
FG113_reverse_unpaired.fq.gz FG35_sorted.bam FGIntype_trimmed_1.fastq.gz KGD465_sorted.bam zygia917_reverse_paired.fq.gz
FG113_reverse_unpaired_fastqc.html FG35_sorted.bam.bai FGIntype_trimmed_1u.fastq KGD465_sorted.bam.bai zygia917_reverse_paired_fastqc.html
FG113_reverse_unpaired_fastqc.zip FG35_trimmed_1.fastq FGIntype_trimmed_2.fastq KGD465_trimmed_1.fastq zygia917_reverse_paired_fastqc.zip
FG113_sorted.bam FG35_trimmed_1.fastq.gz FGIntype_trimmed_2.fastq.gz KGD465_trimmed_1.fastq.gz zygia917_reverse_unpaired.fq.gz
FG113_sorted.bam.bai FG35_trimmed_1u.fastq FGIntype_trimmed_2u.fastq KGD465_trimmed_1u.fastq zygia917_reverse_unpaired_fastqc.html
FG113_trimmed_1.fastq FG35_trimmed_2.fastq Inga_unique_baits.1.bt2 KGD465_trimmed_2.fastq zygia917_reverse_unpaired_fastqc.zip
FG113_trimmed_1.fastq.gz FG35_trimmed_2.fastq.gz Inga_unique_baits.2.bt2 KGD465_trimmed_2.fastq.gz zygia917_sorted.bam
FG113_trimmed_1u.fastq FG35_trimmed_2u.fastq Inga_unique_baits.3.bt2 KGD465_trimmed_2u.fastq zygia917_sorted.bam.bai
FG113_trimmed_2.fastq FGIntype Inga_unique_baits.4.bt2 Ref.fna zygia917_trimmed_1.fastq
FG113_trimmed_2.fastq.gz FGIntype.empties Inga_unique_baits.rev.1.bt2 Ref.fna.fai zygia917_trimmed_1.fastq.gz
FG113_trimmed_2u.fastq FGIntype.vcf Inga_unique_baits.rev.2.bt2 acc zygia917_trimmed_1u.fastq
FG35 FGIntype_1.fastq.gz KGD465 bam_me.sh zygia917_trimmed_2.fastq
FG35.empties FGIntype_1_fastqc.html KGD465.empties cut_out zygia917_trimmed_2.fastq.gz
FG35.vcf FGIntype_1_fastqc.zip KGD465.vcf fastqcfiles zygia917_trimmed_2u.fastq
The file {$acc}_bowtie_output shows the aligment summary for the bowtie2 step. We can take a look inside those file with:
awk 'FNR==1{print "::::\n"FILENAME"\n::::"}1' *_bowtie_output
You can also visually explore the bam files using the software Tablet. You will need the .bam and the index .bai files, which were generated with the bam_me.sh script. Below is an example of what it looks like. It is a good idea to explore the .bam files to check how the reads mapped back to the baits.
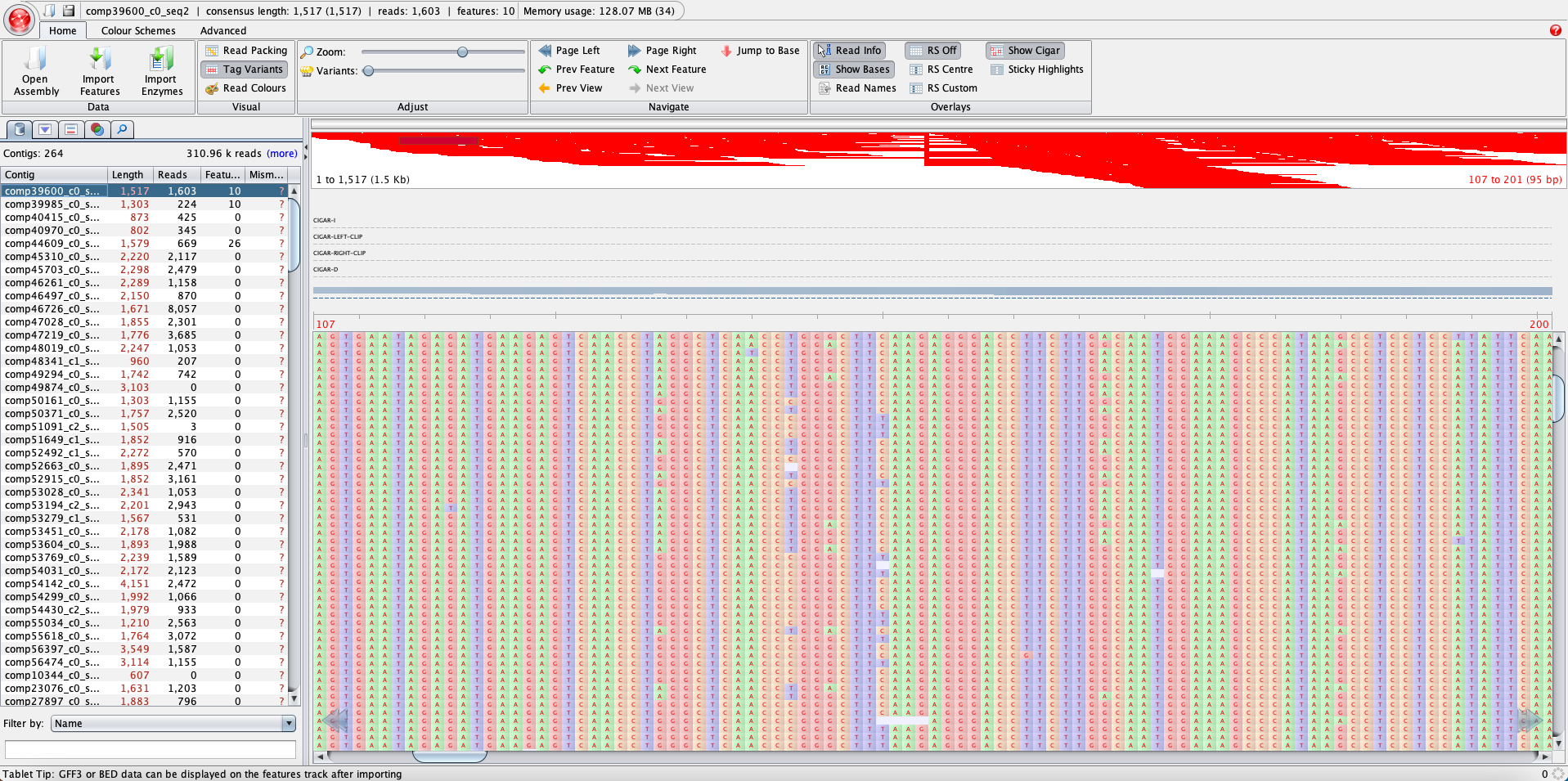
One of the outputs this script produces is the vcf file. VCF is the standard file format for storing variation data.
What are VCF files
The Variant Call Format (VCF) specifies the format of a text file used in bioinformatics for storing gene sequence variations. The format has been developed with the advent of large-scale genotyping and DNA sequencing projects, such as the 1000 Genomes Project. Existing formats for genetic data such as General feature format (GFF) stored all of the genetic data, much of which is redundant because it will be shared across the genomes. By using the variant call format only the variations need to be stored along with a reference genome. More info: http://www.internationalgenome.org/wiki/Analysis/vcf4.0 and http://augustogarcia.me/statgen-esalq/Hapmap-and-VCF-formats-and-its-integration-with-onemap/
We will use the script clean_vcf.sh to edit the vcf files to remove indels and calls with quality less than 36 and then output the consensus fasta. Note that we will need the perl script vcfutils_dasta.pl in the folder we are working on.
clean_vcf.sh
#! /bin/bash -x
# to remove tricky stuff from the vcf files and make the fasta
# Input on comnad line is the accession stem
#Catherine Kidner 11 Nov 2014
echo "Hello world"
acc=$1
echo "You're working on accession $1"
vcf=${acc}.vcf
clean=${acc}_clean.vcf
fasta=${acc}.fasta
grep -v "INDEL" $vcf | awk '{if ($6 >= 36) print $0}' > $clean
perl vcfutils_fasta.pl vcf2fq $clean > $fasta
rm $clean
exit 0
clean_vcf_array.sh
Tip
In case you are doing your anaylisis in Crop Diversity HPC, you can run this step as an array. An array job is a job in which the script is run concomitantly for each sample and it will be much quicker. In this link you can find more about array jobs: https://help.cropdiversity.ac.uk/slurm-overview.html#array-jobs. Below an example of how to run the clean_vcf_array.sh script as an array:
#!/bin/bash
# to remove tricky stuff from the vcf files and make the fasta
# Input on comnad line is the accession stem
# Catherine Kidner 11 Nov 2014, adapted to run in array Flavia Pezzini Mar 2021
#SBATCH --job-name="clean_vcf"
#SBATCH --export=ALL
#SBATCH --mail-user=youremail@yourdomain # enter your email to receive a message once it is done.
#SBATCH --mail-type=END,FAIL
#SBATCH --output ./slurm-%x-%\A_%a.out # %x gives job name, %A job ID, %a array index
#SBATCH --partition=short
#SBATCH --cpus-per-task=4 #number of threads, not cores
#SBATCH --mem=1G #adjust this according to your data.
#SBATCH --array=0-4 # the number of samples you have. We have five accessions we use 0-4 because Bash array is zero-indexed (instead of 1-5). It is a good practice to ask for a maximum of 25 tasks at a time.
acc=$(sed -n "$SLURM_ARRAY_TASK_ID"p /path/to/my/acc/file)
echo "Hello world"
echo "Working on $acc"
vcf=${acc}.vcf
clean=${acc}_clean.vcf
fasta=${acc}.fasta
grep -v "INDEL" $vcf | awk '{if ($6 >= 36) print $0}' > $clean
perl vcfutils_fasta.pl vcf2fq $clean > $fasta
rm $clean
exit 0
To submit just type: sbatch clean_vcf_array.sh
Let’s run the clean_vcf.sh script:
while read f; do ./clean_vcf.sh.sh "$f" ; done < acc
We now have one fasta file per accession, and each file contains the recovered sequence for each of the 264 loci.
We will convert from multifastas of loci per accession to multifasta of accession per locus using the python script switch_multifastas.py. This script needs a folder called By_locus, a list of loci called locus_list and a list of files called fasta_files.
Let’s create the folder and lists:
mkdir By_locus
cat Ref.fna | grep ">" | sed 's/\>//g' > locus_list
ls *.fasta > fasta_files
And then run:
python /path/to/your/Targeted_enrichment/Switch_multifastas.py
Switch_multifastas.py
'''take a set of multifastas and re-order as by filename a set of multifastas
For dealing with hyb_baits bowtie to fasta outputs
needs alist f the fasta file names and a list of the loci (as in the fasta_files)
set at the default - can change to querry for list names
needs an output folder called By_locus'''
def fasta_dict(fastafile):
file = open(fastafile)
name2seq = {}
for line in file:
if line.startswith(">"):
seq = ''
split_line = line.split(' ')
name = split_line[0]
name = name.rstrip()
name = name.lstrip('>')
else:
seq = seq + str(line)
seq = seq.rstrip()
name2seq[name] = seq
return name2seq
print ("Hello world")
fastafile_list = "fasta_files"
locus_list_file = "locus_list"
#fastafile_list = input("Which list of fasta files (full file name please)?\n")
#locus_list_file = input("Which file for names of loci?\n")
#open files for each locus (for reading and writting) ready to feed in the fastas
if locus_list_file != None:
locus_list = open(locus_list_file)
for name in locus_list:
name = name.rstrip("\n")
out_file_name = name + ".fasta"
outfile = open(out_file_name, "w")
outfile.close()
# go through each fasta file pulling out the match to each locus and putting into the file
if fastafile_list != None:
fastafiles = open(fastafile_list)
for file_name in fastafiles:
file_name = file_name.rstrip("\n")
accession = file_name.rstrip(".fna")
#Make a dict of that fasta file
ifasta_dict = fasta_dict(file_name)
keep_lines_processed = 0
keep_seq_found = 0
missing_list = []
if locus_list_file != None:
locus_list = open(locus_list_file)
#for a locus pull out the hit and append to the correct file
for name in locus_list:
name = name.rstrip("\n")
found_fasta = ">" + accession + "_" + name + "\n"
keep_lines_processed += 1
fasta_seq = ifasta_dict.get(name, "NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN")
if name in ifasta_dict.keys():
keep_seq_found +=1
else:
missing_list.append(name)
found_fasta = found_fasta + fasta_seq + "\n"
locus_file_to_write = "By_locus/" + name + ".fasta"
with open(locus_file_to_write, "a") as myfile:
myfile.write(found_fasta)
print("Keep lines processed for " + file_name + " = " + str(keep_lines_processed))
print("Keep sequences found for " + file_name + " = " + str(keep_seq_found))
print("Missing for " + file_name + " = " + str("\n".join(missing_list)))
We now have a folder callled By_locus with 264 fasta files ready to be aligned with mafft.